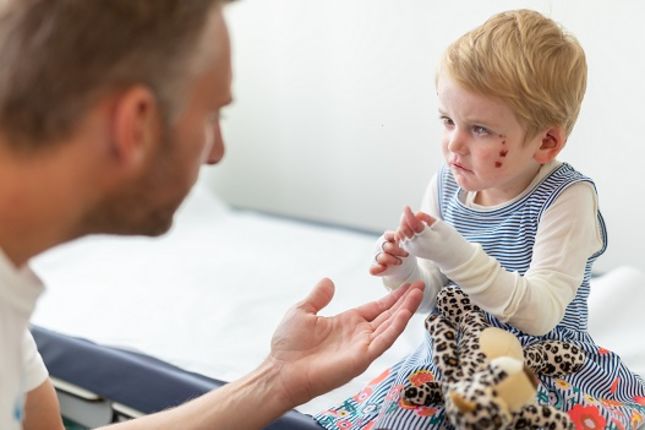
L’épidermolyse bulleuse (EB) est une maladie génétique qui perturbe la façon dont les cellules et les tissus sont maintenus ensemble. Il existe plusieurs formes d’épidermolyse bulleuse mais un symptôme commun à toutes ces différentes formes est une peau extrêmement fragile qui se déchire facilement ou développe des plaies et des cloques en cas de frottement léger, de stress et de blessures mineures. Il n’existe actuellement aucun traitement contre cette maladie, bien que la recherche médicale et certains traitements actuellement en cours d'essais cliniques aient donné des résultats très prometteurs. Comment la thérapie génique et cellulaire pourrait-elle aider?
Que savons-nous?
Toutes les formes d’épidermolyse bulleuse sont causées par des mutations dans des gènes, qui perturbent le codage de protéines importantes pour la connexion des cellules et des tissus. Les quatre principaux types d’épidermolyse bulleuse sont : l’épidermolyse bulleuse simple, l’épidermolyse bulleuse jonctionnelle, l’épidermolyse bulleuse dystrophique et le syndrome de Kindler.
L’épidermolyse bulleuse est relativement rare, son incidence étant estimé à 1 cas pour 50000 naissances.
Visuellement, l’épidermolyse bulleuse affecte la peau mais cette maladie peut également causer des complications dans d’autres organes comme des cloques et des plaies dans la gorge, les voies respiratoires supérieures et les voies urinaires. Elle peut entraîner une maladie systémique progressive dont les conséquences limitent l’éspérance de vie ou conduire à des cancers cutanés agressifs.
Sur quoi les chercheurs se penchent-ils?
De nombreux nouveaux dispositifs, médicaments et traitements biologiques font l'objet d'essais cliniques. Beaucoup de ces études visent à améliorer la prise en charge de l'épidermolyse bulleuse. Quelques études visent à "réparer" les mutations génétiques sous-jacentes à cette maladie.
La recherche visant à développer des thérapies géniquespourrait permettre de restaurer des gènes sains dans les cellules de la peau. Travailler avec des cellules souches de la peau, optimiser les méthodes de culture de la peau en laboratoire et améliorer les méthodes de greffe de tissus sont également des éléments clés dans le développement des thérapies géniques efficaces contre l'épidermolyse bulleuse.
Quels sont les défis à relever?
Le traitement d’une maladie génétique est très difficile car la cause de cette maladie se situe dans l’ADN de la personne. Les traitements visant à compenser les mutations génétiques doivent réparer le gène mutant ou ajouter une nouvelle copie fonctionnelle de ce gène dans les cellules. Cette tâche n’est pas simple. Modifier l’ADN des cellules ou ajouter de nouveaux gènes dans les cellules comportent des risques potentiels, comme la possibilité d’induire un cancer. Les greffes de cellules ou de peau peuvent comporter des risques comme des saignements, des infections ou le rejet du greffon.
Un autre défi consiste à rendre les traitements complexes, comme la thérapie génique, abordable et accessible à tous.
A propos de l’épidermolyse bulleuse
L’épidermolyse bulleuse désigne un groupe de maladies génétiques rares du tissu conjonctif qui provoquent une peau très fragile et des cloques. Les cloques peuvent être causées par le frottement de la peau, des blessures mineures et des activités quotidiennes, comme le frottement et le grattage. Ces cloques peuvent devenir des blessures douloureuses et des plaies ouvertes. Dans les formes graves de cette maladie, les cloques et les plaies se développent également dans d’autres tissus, comme dans la bouche, la gorge, l’estomac, les voies respiratoires supérieures et les voies urinaires.
La gravité de l’épidermolyse bulleuse est très variable. Les formes légères de cette maladie peuvent entrainer le traitement de cloques mineures tout au long de la vie, tandis que d’autres ne présentent que des symptômes sévères pendant l’enfance qui peuvent s’améliorer avec l'âge. D'autres formes d'épidermolyse bulleuse peuvent présenter un risque vital en raison de la perte de grandes quantités de peau. D'autres peuvent provoquer des cicatrices étendues, la fusion des doigts et des orteils ainsi que d'autres complications limitant la durée de vie. Certaines formes de la maladie peuvent entraîner le développement d'un cancer de la peau, réduisant ainsi l'espérance de vie.
L’épidermolyse bulleuse est rare, ce qui rend difficile l'estimation du nombre total de personnes atteintes de cette maladie. DEBRA, une organisation caritative consacrée à l'épidermolyse bulleuse, estime qu'environ 500 000 personnes dans le mondesont atteintes d'une forme de cette maladie.
Toutes les formes héréditaires d'épidermolyse bulleuse sont causés par des mutations dans l'ADN. Ces mutations perturbent l'un des gènes importants pour les protéines qui relient les cellules et les tissus entre eux. C'est pourquoi cette maladie est également considérée comme une maladie du "tissu conjonctif". Actuellement, 16 gènes de notre ADN ont été associés à l'épidermolyse bulleuse héréditaire classique.
Une mutation génétique liée à l'épidermolyse bulleuse est souvent transmise des parents aux enfants. Notre ADN contient deux copies de chaque gène (à l'exception des gènes situés sur les chromosomes X et Y). Pour plusieurs formes d'épidermolyse bulleuse, il suffit d'hériter d'une seule copie d'une mutation génétique liée à la maladie. C'est ce qu'on appelle une forme "dominante" d'épidermolyse bulleuse. Le parent qui transmet une forme dominante de mutation génétique liée à l'épidermolyse bulleuse est également atteint de la maladie et de ses symptômes. D'autres formes d'épidermolyse bulleuse nécessitent d'hériter de la mutation du gène lié à l'épidermolyse bulleuse des deux parents. On parle alors de formes "récessives" d'épidermolyse bulleuse. Les formes récessives d'épidermolyse bulleuse sont souvent une surprise pour la famille, car aucun des deux parents ne présente les symptômes de la maladie et peut ne pas savoir qu'il est porteur de la mutation.
A propos de la peau
Notre peau est un organe vaste et complexe. Cet organe agit comme un bouclier imperméable qui garde l’eau à l’intérieur et les bactéries à l’extérieur. Elle nous protège du vent et des intempéries et nous aide à réguler notre température corporelle. C’est là que la vitamine D est synthétisée.
De plus, notre peau possède un grand nombre de cellules spécialisées qui nous permettent de percevoir la température, les textures, la pression et la douleur. Notre peau est faite pour durer, du moins si tout se passe comme prévu.
Les deux principales couches de la peau sont l’épiderme (la couche la plus externe) et le derme. Les chercheurs subdivisent ensuite ces couches en couches (ou strates) spécifiques de cellules. Vous pouvez voir ces sous-couches dans le diagramme.
L’épiderme est constitué principalement de cellules appelées kératinocytes. Elles doivent leur nom à la protéine résistante qu’elles produisent : la kératine - il s’agit de la même protéine que celle qui compose nos cheveux et nos ongles. Les kératinocytes sont produits par les cellules souches de la peau qui résident dans la sous-couche basale de l'épiderme et dans les follicules pileux du derme. Les cellules souches de la peau fabriquent constamment de nouveaux kératinocytes pour remplacer les cellules de la peau qui tombent continuellement. En fait, presque toutes les cellules de la couche externe de la peau sont remplacées toutes les 4 à 5 semaines ! Les cellules souches cutanées sont donc très importantes pour le maintien de la santé de notre peau.
Le derme est la couche de la peau qui contient les vaisseaux sanguins, les cellules sensorielles du toucher, de la température et de la douleur, les glandes sudoripares, les follicules pileux, etc. Il existe de nombreux types de cellules dans le derme, mais la majeure partie du derme est en fait constituée d'un maillage souple de fibres conjonctives et de protéines. Ce maillage de protéines soutient la structure de la peau et maintient le tout en place. Les protéines impliquées dans ce maillage sont principalement le collagène et l'élastine.
Le point de jonction entre l'épiderme et le derme est également important, notamment lorsqu'on parle d'épidermolyse bulleuse. Ce point de jonction, appelé "membrane basale", est une fine couche de tissu conjonctif qui joue un rôle important dans la liaison entre l'épiderme et le derme. La membrane basale est principalement constituée de deux types de protéines, la laminine et le collagène.
Illustration 1: Peau saine
Les quatre formes principales d’épidermolyse bulleuse
Il existe quatre formes principales d’épidermolyse bulleuse : l’épidermolyse bulleuse simple, l’épidermolyse bulleuse jonctionnelle, l’épidermolyse bulleuse dystrophique et le syndrome de Kindler. Ces catégories sont généralement distinguées par le point de séparation entre la couche externe de la peau (l'épiderme) et le derme (couche inférieure de la peau).
L’épidermolyse bulleuse simple
L’épidermolyse bulleuse simple est la forme la plus courante d’épidermolyse bulleuse, représentant environ 70% de tous les cas. Dans ce type d’épidermolyse bulleuse, la séparation de l'épiderme et du derme se produit au niveau de la couche basale des cellules de l'épiderme (Strate basale), juste au-dessus de la membrane basale. L’épidermolyse bulleuse simple est principalement héritée des parents sous la forme d’un caractère dominant, ce qui signifie qu'un seul parent doit transmettre la mutation du gène lié à la maladie. Ce parent est atteint d'épidermolyse bulleuse simple et présente probablement (ou a probablement présenté) les symptômes associés.
La gravité des symptômes de l'épidermolyse bulleuse simple peut varier, allant de la fragilité de la peau et des petites cloques sur les mains et les pieds, aux cas où les cloques se produisent sur tout le corps, jusqu’à des sous-types plus graves. Les formes légères peuvent s'améliorer avec l'âge, mais la plupart des individus sont confrontés à des cloques tout au long de l'enfance et à l'âge adulte.
L’épidermolyse bulleuse jonctionnelle (EBJ)
L’épidermolyse bulleuse jonctionnelle représente environ 10% de tous les cas d’épidermolyse bulleuse. Dans ce type d’épidermolyse bulleuse, la séparation de l'épiderme et du derme se produit au niveau de la membrane basale. L’épidermolyse bulleuse jonctionnelle est transmise par les parents sous la forme d'un caractère récessif, ce qui signifie qu'un individu doit avoir hérité d'une mutation génétique liée à l'épidermolyse bulleuse de ses deux parents. Ces parents ne présentaient probablement aucun symptôme de la maladie.
Certaines personnes touchées par l’épidermolyse bulleuse jonctionnelle auront une “épidermolyse bulleuse jonctionnelle sévère" (appelé auparavant épidermolyse bulleuse jonctionnelle sévère généralisée ou épidermolyse bulleuse jonctionnelle de Herlitz). Les cloques sont présentes sur tout le corps, y compris la bouche, le nez et la gorge. Ces cloques empêchent les nouveau-nés de manger et de respirer correctement, ce qui entraîne une malnutrition et une insuffisance pulmonaire. Tragiquement, ce sous-type extrême de la maladie présente un risque vital, et très peu d'enfants atteints d’épidermolyse bulleuse jonctionnelle sévère vivent au-delà de deux ans.
L'autre grand sous-type est "l’épidermolyse bulleuse jonctionnelle intermédiaire" (précédemment appelé épidermolyse bulleuse jonctionnelle intermédiaire généralisée ou épidermolyse bulleuse jonctionnelle non-Herlitz). Les symptômes de ce groupe sont moins graves, mais toujours intenses. Des cloques apparaissent sur tout le corps, y compris dans la bouche, le nez et la gorge. Malheureusement, les décès sont encore fréquents avec cette forme d'épidermolyse bulleuse jonctionnelle. Les personnes atteintes d'épidermolyse bulleuse jonctionnelle intermédiaire peuvent atteindre l'âge adulte, mais elles présentent souvent de grandes cicatrices, des problèmes d'ongles et de dents, ainsi que d'autres complications qui limitent leur durée de vie.
L’épidermolyse bulleuse dystrophique (EBD)
L’épidermolyse bulleuse dystrophique représente environ 20 % de tous les cas d'épidermolyse bulleuse. Dans le cas de l'épidermolyse bulleuse dystrophique, la séparation de l'épiderme et du derme se produit sous la membrane basale. Cette maladie peut être héritée sous la forme d’un caractère dominant ou récessif, en fonction de la mutation du gène lié à l'épidermolyse bulleuse.
La gravité des symptômes varie considérablement selon les cas. Un trait commun est que les cloques entraînent des cicatrices. De nombreux cas causés par la forme dominante de l'épidermolyse bulleuse présentent des ampoules et d'autres symptômes légers, ce qui permet à ces personnes de mener une vie avec une légère déficience. La plupart des personnes atteintes de la forme récessive de l'épidermolyse bulleuse doivent faire face à beaucoup plus de limitations, car les cloques et les cicatrices sont beaucoup plus graves, et le risque de cancer de la peau est plus élevé. Parmi les autres symptômes possibles, citons le défigurement lors de la cicatrisation des ampoules et des plaies, la fusion des doigts et des orteils, la limitation des mouvements des articulations et le rétrécissement de l'œsophage (tube alimentaire), qui rend la déglutition difficile.
Le Syndrome de Kindler (SdK)
Le syndrome de Kindler est un sous-type rare d'épidermolyse bulleuse héréditaire dans lequel des cloques peuvent apparaître dans différentes couches de la peau. Le syndrome de Kindler est une forme récessive d'épidermolyse bulleuse, ce qui signifie qu'un individu hérite de la mutation génétique de ses deux parents. Les symptômes peuvent aller de légers à graves, et comprennent une fragilité de la peau dès la naissance, une sensibilité à la lumière, une décoloration et un épaississement de la peau. Les maladies des gencives, les ampoules buccales et les intestins enflammés peuvent affecter l'alimentation, et les personnes atteintes de ce sous-type ont un risque accru de développer un cancer de la peau.
Nos partenaires du réseau de recherche de l’épidermolyse bulleuse ont des informations sur l’épidermolyse bulleuse et sur sa classification.
Quel est le problème dans l’épidermolyse bulleuse?
Bien que toutes les formes d’épidermolyse bulleuse aient des cloques comme symptôme, elles ne sont pas causées par le même problème. Le gène affecté par une mutation génétique lié à cette maladie détermine l’endroit, la raison et l’ampleur de la formation de cloques et du décollement de la peau. Il détermine également le type d’épidermolyse bulleuse dont une personne est atteinte.
Actuellement, il existe 16 gènes spécifiques dans notre ADN qui ont été mise en cause dans l’épidermolyse bulleuse classique. Les mutations dans ces gènes empêchent les cellules de fabriquer des protéines fonctionnelles qui maintiennent l'épiderme et le derme ensemble. Savoir quel rôle joue normalement les protéines pour maintenir la peau en place est essentiel pour comprendre les différences entre l'épidermolyse bulleuse simple, l’épidermolyse bulleuse jonctionnelle, et l’épidermolyse bulleuse dystrophique. Cela révèle également pourquoi certains types d'épidermolyse bulleuse sont plus graves que d'autres et pourquoi un traitement pour un type d’épidermolyse bulleuse peut ne pas fonctionner pour tous.
Vous trouverez ci-dessous des résumés qui mettent en évidence les gènes, leurs protéines et certains détails sur le lieu et la raison du détachement pour trois des principales formes d'épidermolyse bulleuse.
L’épidermolyse bulleuse simple (EBS)
Dans ce type d’épidermolyse bulleuse, la séparation de l'épiderme et du derme se produit au niveau de la couche basale des cellules de l'épiderme (Strate basale), juste au-dessus de la membrane basale.
Le problème est que les protéines qui fixent les cellules basales à la membrane basale ne se lient pas correctement au support structurel de la cellule (le cytosquelette). Les cellules basales se déchirent alors et se rompent, laissant la partie inférieure de ces cellules attachée à la membrane basale.
Les principales protéines affectées par les mutations génétiques dans l'épidermolyse bulleuse simple sont les suivantes :
Kératine de type 5 - [Nom du gène : KRT5]
Kératine de type 14 - [Nom du gène : KRT14]
Plectine - [Nom du gène : PLEC1]
Illustration 2 : peau saine et peau en épidermolyse bulleuse simple
L’épidermolyse bulleuse jonctionnelle (EBJ)
Cette maladie se caractérise par une séparation de l’épiderme et du derme au niveau de la membrane basale, spécifiquement dans la sous-couche “lamina lucida”. Cette sous-couche est constituée de protéines qui ancrent les cellules basales de l’épiderme au derme. Quand certaines protéines sont manquantes ou dysfonctionnelles, cette jonction ne permet pas de maintenir ensemble l’épiderme et le derme.
Les principales protéines affectées par des mutations dans les EBJ sont:
Collagène de type XVII (ou BPAG2) [Nom du gène: COL17A1]
Intégrine a6b4 (sous-unités alpha 6 et beta 4) - [Nom des gènes: ITGA6 et ITGA4]
Laminine-332 (ou Laminine-5) - [Nom des gènes: LAMA3, LAMB3 et LAMC2]
Illustration 3: Peau saine et peau en épidermolyse bulleuse jonctionnel (EBJ)
L’épidermolyse bulleuse dystrophique (EBD)
Dans l’EBD, la séparation de l’épiderme et du derme se produit sous la membrane basale. La principale protéine qui lie cette membrane au derme est le collagène VII. Cette protéine forme des fibrilles d’ancrage qui se tissent dans les protéines structurelles du derme. Sans le collagène VII, la membrane basale se détache facilement du derme.
La protéine généralement affectée par une mutation génétique dans l’EBD est:
Collagène de type VII - [Nom du gène: COL7A1]
Illustration 4: Peau saine et peau en épidermolyse bulleuse dystrophique (EBD)
Le Syndrome de Kindler (SdK)
Le SdK peut affecter différentes couches de la peau. L’absence ou le dysfonctionnement d’une protéine empêche les kératinocytes (cellules de la peau) de proliférer et se diviser convenablement, ainsi que d’attacher l’épiderme au derme.
La principale protéine affectée d’une mutation dans l’EBK est:
Kindline-1 [Nom du gène: FERMT1]
Traitements de l’EB
Il n’y a actuellement pas de thérapies approuvées qui corrigent la mutation génétique causale de l’EB. Les traitements actuels se concentrent principalement sur la prévention et le soulagement des symptômes de l’EB. Les mesures préventives impliquent l’utilisation de rembourrages et de couches protectrices pour minimiser la friction et autres traumatismes mineurs de la peau. Les traitements visent à prévenir les infections et aider dans la guérison des cloques et des plaies.
Dans les cas plus sévères d’EB, des médicaments peuvent être utilisées pour éviter l’infection des lésions et plaies ouvertes. Une intervention chirurgicale peut également être effectuée pour empêcher les doigts et les orteils de fusionner ou que la gorge et l'œsophage (tuyau alimentaire) ne deviennent trop étroits.
Dans les cas extrêmes d’EB, les médecins peuvent couvrir de grandes plaies ouvertes en procédant à des greffes de peau. Cependant, il est très rare de greffer de la peau de donneurs car le système immunitaire la rejettera presque toujours, même en cas de proche parenté entre donneur et receveur. Ainsi, les médecins utilisent souvent des substituts cutanés temporaires pour couvrir les grandes plaies ouvertes. De nouveaux “substituts cutanés biologiques” sont également de plus en plus utilisés.
Recherches en cours
Amélioration de la qualité de vie
Il n’existe actuellement aucun traitement cliniquement approuvé qui “répare” la cause biologique sous-jascente de l’EB. Cependant, de nombreux chercheurs, sociétés pharmaceutiques et sociétés de fournitures médicales travaillent à développer de meilleurs produits pour améliorer la gestion des symptômes de l'EB.
Les produits considérés peuvent aller de bandages plus adaptés à des crèmes régénératrices pour la peau. Plusieurs essais cliniques sont en cours pour différents types de pansements et crèmes pour voir s’ils peuvent accélérer le processus de guérison de la peau chez les personnes atteintes d’EB.
Dans les cas sévères d’EB, les médecins peuvent avoir besoin de couvrir de grandes plaies ouvertes. Cela implique souvent de les couvrir avec des substituts de peau synthétiques. Cependant, des entreprises ont récemment mis au point des « substituts cutanés biologiques » plus avancés, tels que Biobrane®, Integra®, Orcel®, Apligraft® et autres. Ces substituts cutanés avancés visent à améliorer la cicatrisation des plaies en utilisant des mailles synthétiques sur lesquelles les cellules se développent mieux, des protéines présentes naturellement dans la peau ou parfois même des kératinocytes (cellules de la peau) vivants. Ce ne sont que quelques exemples de comment les technologies et produits en cours de développement peuvent grandement aider à améliorer la qualité de vie des personnes atteintes d’EB.
Comment refaire une ‘nouvelle’ peau
Le traitement idéal pour une maladie génétique est de corriger ou remplacer la mutation du gène causale dans l’ADN de la personne. Pour ce faire, chercheurs ont crée des “thérapies géniques”. Ces dernières années, plusieurs essais cliniques et traitements de ce genre ont réussi, ce qui indique qu’il pourrait bientôt y avoir des traitements qui s’attaquent à la cause de l’EB pour la première fois.
Les thérapies géniques sont actuellement dans les premières phases de développement. De nouvelles découvertes et technologies, telles que l’édition de gènes par CRISPR/Cas9, ont considérablement accéléré les méthodes d’édition de gènes. Cependant, changer l’ADN dans les cellules d’une personne n’est pas simple et peut entraîner de sérieux risques. Si l’édition ou l’addition d’un gène tourne mal, cela peut causer un cancer, ajouter de nouvelles complications, ou empirer la maladie d’une personne. Les essais cliniques sont cruciaux pour identifier les risques posés par les nouveaux traitements et pour prouver leur efficacité.
Souvent, les cellules souches vont de paire avec les thérapies géniques. Pour les thérapies géniques liées à l’EB, les chercheurs veulent éditer l’ADN des cellules souches cutanées plutôt que d’autres cellules de la peau. Cela est important pour que l’addition (ou édition) d’un gène soit transmise aux autres cellules de l’épiderme descendant de la cellule souche, puisque presque tous les kératinocytes de notre peau sont remplacés toutes les 4-5 semaines. De plus, les cellules souches cutanées se renouvellent continuellement, donc l’édition d’ADN dans ces cellules pourrait être ‘permanent’ (au moins pour la durée de vie de ces cellules souches).
Plusieurs traitements en cours d’essai clinique testent différentes méthodes d’édition de l’ADN des cellules souches de la peau des patients afin de les utiliser pour générer de la ‘nouvelle’ peau saine pour la greffer. Même si les méthodes et détails de ces essais diffèrent, l’idée et le processus de base restent les mêmes. Il s’agit de prendre un échantillon de peau d’une personne touchée par l’EB et de cultiver ces cellules en laboratoire en espérant que cet échantillon contienne des cellules souches cutanées. Des méthodes d’édition d’ADN sont ensuite utilisées pour ajouter ou éditer des gènes afin de rétablir une production normale de protéines. Les cellules contenant les modifications d’ADN voulues sont ensuite utilisées dans des méthodes avancées de culture cellulaire pour obtenir des couches de peau en laboratoire. Celles-ci peuvent ensuite être greffées sur le patient. Certaines de ces approches utilisent des cellules souches pluripotentes induites (CSPi), un type de cellule souche qui peut être généré à partir de cellules de notre corps, qui prolifère indéfiniment et qui peut générer tous les types de cellules de la peau. Elles peuvent être une source de cellules pour remplacer les tissus endommagés. Bien que les CSPi soient approuvées dans plusieurs essais cliniques, les patients qui reçoivent des transplantations de ces cellules pourraient avoir un risque accru de développer un cancer.
Ce type de traitement requiert beaucoup de temps et d’effort pour identifier le problème génétique pour chaque personne atteinte d’EB, développer une méthode spécifique d’édition d’ADN, des semaines pour faire pousser les cellules et des chirurgiens pour greffer la nouvelle peau. Ces traitements nécessitent également des installations de recherche et de culture cellulaire très spécialisés. Malgré ces obstacles, des avancées majeures ont prouvé que tout cela est possible: la section suivante explique l’un de ces succès.
Zoom sur la recherche en Europe: une histoire à succès
En 2017, un groupe de chercheurs et de cliniciens dirigé par le professeur Michele De Luca a rapporté avoir utilisé avec succès la thérapie génique et des greffes de peau cultivées en laboratoire pour sauver un garçon à qui il manquait plus de 80 % de sa peau. Le garçon souffrait d’EBJ causé par des mutations du gène LAMB3 qu'il avait hérité de ses deux parents. Tous les traitements, y compris plusieurs approches extrêmes, avaient échoué et les cliniciens pensaient que le garçon avait peu de chances de survivre. Les autorités gouvernementales ont accordé une autorisation pour “usage compassionnel” d'un traitement préliminaire de thérapie génique qui n'avait été testé auparavant que dans deux études de cas portant chacune sur un seul patient. Les parents du garçon ont également accepté d'essayer la procédure, même après avoir été informés que le garçon pourrait ne pas survivre à la procédure elle-même.
Les chercheurs ont commencé par collecter des échantillons de peau du garçon et à cultiver ces cellules en laboratoire. Ils espéraient obtenir des amas de cellules contenant des cellules souches cutanées. Les amas de cellules ont ensuite été traités avec un virus pour ajouter une copie fonctionnelle du gène LAMB3 à l'ADN des cellules souches cutanées. Ce processus ne résout pas la mutation que le garçon avait dans ses propres copies du gène, mais ajoute au contraire de l’ADN avec une copie du gène LAMB3 aux cellules souches de la peau du garçon. Ce nouveau gène permet aux cellules de fabriquer des protéines de laminine fonctionnelles. Les cellules souches cutanées avec le gène LAMB3 fonctionnel ajouté ont ensuite été utilisées pour faire pousser des couches de peau en laboratoire à l'aide de méthodes de culture avancées. Ce processus prend plusieurs semaines. Les petits morceaux de peau cultivés en laboratoire ont ensuite été greffés sur le garçon lors de deux procédures distinctes à un mois d'intervalle. En fin de compte, le garçon a survécu et vit avec une peau constituée de cellules modifiées par l'ADN sur 80 % de son corps. Les chercheurs ont examiné cette peau et disent qu'elle ressemble remarquablement à une peau normale à bien des égards.
Ce succès est très enthousiasmant, et certains diraient même miraculeux car il a dépassé les attentes. Malheureusement, les traitements de thérapie génique comme ceux-ci ne sont pas encore cliniquement approuvés ou largement disponibles. Il est également important de réaliser que ces thérapies géniques doivent « corriger » les problèmes causés par des gènes mutés spécifiques de chaque patient. Une médecine personnalisée comme celle-ci coûte cher, nécessite des installations spécialisées et le travail de nombreuses personnes. Espérons que des traitements comme celui-ci deviendront approuvés cliniquement, abordables et largement disponibles pour de nombreuses formes d'EB.
L'avenir s’annonce prometteur. Le succès décrit ci-dessus, ainsi que les résultats positifs d’autres études, ont abouti sur de nouvelles études cliniques. M. De Luca du centre de Médecine Régénérative “Stefano Ferrari” à Modène, en Italie, et JW Bauer de l’hôpital universitaire de dermatologie de Salzbourg en Autriche, mènent plusieurs études cliniques utilisant des procédures similaires mais plus avancées pour remplacer les mutations du gène COL7A1 et COL17A1 (étude 1, étude 2). Des chercheurs d’autres institutions développent également leurs propres thérapies géniques, tel que l’essai clinique mené par Jean Yuh Tang à l’université de Stanford, qui vise également à corriger les mutations du gène COL7A1 dans l’EBD.
Nous espérons que la recherche mènera à des traitements sûrs, efficaces et abordables pour l’EB.
Autres approches thérapeutiques
Comme indiqué dans la section précédente, il n’est pas aisé de fabriquer une peau à ADN modifié: le processus est complexe, coûteux et risqué. De plus, il faudra peut-être des années avant que cette approche ne devienne un traitement réaliste.
Ces défis ont incité les chercheurs à développer d’autres approches de traitement de l’EB qui serait plus rapides, approuvés cliniquement et financièrement accessible aux patients atteints d’EB. Celles-ci impliquent des cellules souches, des médicaments, des extraits de plantes et de nombreux autres trainements. Les deux approches présentées ci-dessous ne sont que des exemples.
Des crèmes et des lotions thérapeutiques locales sont en cours de développement pour contrer les problèmes de mutations spécifiques de l'EB. Deux études cliniques particulièrement intéressantes utilisent des crèmes qui tentent de fixer ou de remplacer le gène COL7A1, ce qui aiderait les personnes atteintes d’EBD. Une crème contient des composants qui aident les cellules de la peau à ignorer la mutation du gène COL7A1 lors de la fabrication de la protéine de collagène VII. L'autre crème contient des particules virales qui insèrent une copie saine du gène COL7A1 dans les cellules. (Ces particules virales ne peuvent pas se répliquer, ce qui les rends sûres à utiliser).
Les greffes de moelle osseuse sont traditionnellement associées à la leucémie et à d'autres troubles du sang et du système immunitaire. Cependant, certains groupes de recherche explorent le traitement de l'EB avec des greffes de moelle osseuse allogéniques, où les cellules souches proviennent de donneurs sains. L'avantage de cette approche est que les greffes de moelle osseuse sont utilisées avec succès depuis de nombreuses années et que la technique s'est grandement améliorée au cours du temps. Des résultats préliminaires suggèrent que les cellules souches saines de la moelle osseuse (ou les cellules fabriquées par ces cellules souches) produisent des protéines du tissu conjonctif qui manquent dans la peau, comme le collagène VII chez les personnes atteintes de DEB. Cependant, cette approche a également ses limites, notamment la nécessité d'un donneur humain immuno-compatible, et les risques associés à cette procédure sont actuellement élevés.
Une liste plus complète des essais cliniques pour l'EB peut être trouvée sur clinicaltrials.gov. (Veuillez noter que ce site Web public ne fait que répertorier les essais cliniques. Il ne vérifie pas si les essais cliniques répertoriés sont sûrs, scientifiquement fondés ou effectués par des institutions crédibles. Veuillez lire tous les avertissements, parler à des professionnels de santé de confiance et vous renseigner sur les risques et les avantages potentiels).