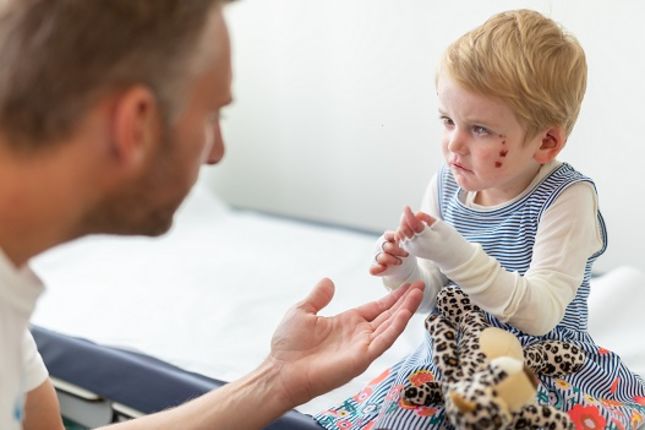
Pęcherzowe oddzielanie się naskórka (Epidermolysis Bullosa - EB) jest zaburzeniem genetycznym, które zakłóca sposób, w jaki komórki i tkanki są razem utrzymywane. Istnieją różne formy EB, ale wspólnym objawem wszystkich rodzajów EB jest wyjątkowo delikatna skóra, która łatwo uszkadza się, ulega zranieniu i ma skłonność do tworzenia pęcherzy nawet z powodu lekkiego tarcia lub drobnych urazów. Obecnie nie ma terapii na EB, aczkolwiek badania medyczne i niektóre metody leczenia testowane w badaniach klinicznych przyniosły obiecujące wyniki. Jak może pomóc terapia genowa i komórkowa?
Co wiemy?
Wszystkie formy EB są spowodowane mutacjami w genach, zaburzającymi wytwarzanie białek ważnych dla łączenia się komórek i tkanek. Cztery podstawowe typy EB to postać zwykła EB, graniczna EB, dystroficzna EB i zespół Kindlera.
EB występuje stosunkowo rzadko: szacunkowa częstość występowania tej choroby wynosi 1 na 50 000 żywych urodzeń.
EB w widoczny sposób wpływa na kondycję skóry, ale zaburzenie to może również powodować powikłania w innych narządach, takie jak pęcherze i rany w gardle, górnych drogach oddechowych oraz drogach moczowych. EB może powodować postępującą chorobę ogólnoustrojową przyczyniając się do skrócenia życia, może także prowadzić do agresywnych nowotworów skóry.
Co badają naukowcy?
W badaniach klinicznych analizowanych jest wiele nowych rozwiązań, leków i terapii biologicznych. Większość z tych badań ma na celu poprawę komfortu życia chorych na EB. Pozostałe badania mają na celu „naprawienie” mutacji genetycznych będących przyczyną EB.
Celem potencjalnych terapii genowych jest możliwość przywrócenia prawidłowej formy genów w komórkach skóry. Praca z komórkami macierzystymi skóry, optymalizacja hodowli skóry w laboratoriach i ulepszanie metod przeszczepiania tkanek są ważnymi zagadnieniami w opracowywaniu skutecznych terapii genowych EB.
Jakie są wyzwania?
Leczenie choroby genetycznej jest bardzo trudne, ponieważ przyczyna leży w DNA danej osoby. Leczenie kompensujące mutacje genów musi naprawić zmutowany gen lub dodać do komórek nową funkcjonalną kopię genu. To nie jest proste zadanie. Modyfikowanie DNA komórki lub dodawanie nowych genów do komórek niesie ze sobą potencjalne ryzyko, na przykład wywołania zmian nowotworowych. Przeszczepy komórek i skóry mogą wiązać się z ryzykiem takim, jak krwawienie, infekcja lub odrzucenie przeszczepu.
Innym wyzwaniem jest również cena i dostępność złożonych metod leczenia takich, jak terapie genowe.
O pęcherzowym oddzielaniu się naskórka
Pęcherzowe oddzielanie się naskórka (Epidermolysis Bullosa - EB) należy do grupy rzadkich genetycznych chorób tkanki łącznej, które są przyczyną powstawania bardzo delikatnej, mającej skłonność do powstawania pęcherzy skóry. Powstawanie pęcherzy może być spowodowane tarciem, drobnymi urazami skóry oraz codziennymi czynnościami, takimi jak drapanie. Pęcherze mogą stać się bolesnymi i otwartymi ranami. W ciężkich postaciach EB pęcherze i rany rozwijają się również w innych elementach ciała, takich jak jama ustna, gardło, żołądek, górne drogi oddechowe lub drogi moczowe.
Objawy EB mogą mieć bardzo różne nasilenie. Łagodne typy EB są przyczyną powstawania drobnych pęcherzy przez całe życie, a inne rodzaje EB wywołują dotkliwe objawy u dzieci, jednak z wiekiem mogą zanikać. Inne formy EB mogą zagrażać życiu z powodu utraty dużej ilości skóry. Jeszcze inne mogą powodować rozległe blizny, zrosty palców rąk i nóg, a także inne komplikacje ograniczające życie. Niektóre formy choroby mogą prowadzić do rozwoju raka skóry, skracając długość życia.
EB jest chorobą rzadką, co sprawia, że oszacowanie całkowitej liczby chorych jest wyzwaniem. DEBRA, organizacja charytatywna poświęcona EB, szacuje, że około 500 000 ludzi na całym świecie ma jedną z form EB.
Wszystkie dziedziczne typy EB są spowodowane mutacjami w DNA danej osoby. Mutacje te zmieniają jeden z kilku ważnych genów kodujących białka uczestniczące w tworzeniu połączeń między komórkami w tkankach. Dlatego EB jest również określany jako zaburzenie „tkanki łącznej”. Obecnie w naszym DNA znajduje się 16 genów, które zostały powiązane z klasycznymi dziedzicznymi postaciami EB.
Mutacja genu powiązana z EB jest często przekazywana dzieciom przez rodziców. Nasze DNA zawiera dwie kopie każdego genu (z wyjątkiem genów na chromosomach X i Y).
W przypadku niektórych postaci EB do wystąpienie choroby wystarczy odziedziczenia tylko jednej kopii mutacji genu sprzężonego z EB. Jest to określane jako „dominująca” forma EB. Rodzic, który przekazuje dominującą formę mutacji genu sprzężonego z EB, ma również EB i jego objawy. Inne formy EB rozwijają się w wyniku odziedziczenia mutacji genu sprzężonego z EB od obojga rodziców. Są to tak zwane „recesywne” formy EB. Recesywne formy EB są często zaskoczeniem dla rodziny, ponieważ żaden z rodziców nie ma objawów EB i może nie wiedzieć, że jest nosicielem mutacji.
Linki do innych stron internetowych użytych w tekscie (dostępne w języku angielskim):
Nasza skóra to duży i złożony narząd. Narząd ten działa jak wodoodporna osłona, utrzymując wodę i bakterie na zewnątrz. Chroni nas przed wiatrem i pogodą oraz pomaga regulować temperaturę ciała. To tam syntetyzowana jest witamina D. Co więcej, nasza skóra ma ogromną liczbę wyspecjalizowanych komórek, które pozwalają nam odczuwać temperaturę, teksturę, ucisk i ból.
Nasza skóra jest wytrzymała, jeśli funkcjonuje w prawidłowy sposób.
Dwie główne części skóry to naskórek (część najbardziej zewnętrzna) i skóra właściwa. Naukowcy wyróżniają w obu tych częściach skóry określone warstwy komórek. Możesz zobaczyć te warstwy na diagramie.
Naskórek składa się głównie z komórek zwanych keratynocytami. Nazwa ta pochodzi od wytwarzanego przez nie białka zwanego keratyną – jest to to samo białko, z którego powstają nasze włosy i paznokcie. Keratynocyty są tworzone przez komórki macierzyste skóry, które znajdują się w warstwie podstawnej naskórka oraz w mieszkach włosowych skóry właściwej. Komórki macierzyste skóry nieustannie wytwarzają nowe keratynocyty, które zastępują nieustannie złuszczane komórki. Prawie wszystkie komórki w zewnętrznej warstwie skóry są wymieniane co 4-5 tygodni! To sprawia, że komórki macierzyste skóry są bardzo ważne dla utrzymania jej zdrowia.
Skóra właściwa to część skóry, która zawiera naczynia krwionośne, komórki czuciowe dotyku, temperatury i bólu, gruczoły potowe, mieszki włosowe i inne. W skórze właściwej występuje wiele różnych typów komórek, ale większość skóry właściwej składa się z elastycznej siatki białek. Ta siatka białek wspiera strukturę skóry i utrzymuje wszystkie jej elementy na właściwym miejscu. Białka zaangażowane w tworzenie tej siatki to głównie kolagen i elastyna.
Ważne jest również miejsce połączenia naskórka ze skórą właściwą, szczególnie przy omawianiu pęcherzowego oddzielania się naskórka. Miejsce to nazywane jest „błoną podstawną” i jest cienką warstwą, ważną dla połączenia naskórka i skóry właściwej. Błona podstawna składa się głównie z dwóch rodzajów białek: lamininy i kolagenu.
Rodzaje te są rozróżniane na podstawie punktu, w którym zewnętrzna część skóry (naskórek) oddziela się od skóry właściwej (dolna część skóry).
Postać zwykła EB (EBS)
Postać zwykła EB (EBS) jest najczęstszą formą EB, stanowiącą około 70% wszystkich przypadków EB. W EBS oddzielenie naskórka od skóry właściwej następuje w warstwie podstawnej komórek naskórka, tuż nad błoną podstawną. EBS jest głównie dziedziczona po rodzicach jako cecha dominująca, co oznacza, że do jej rozwoju wystarczy przekazanie mutacji genu sprzężonego z EB tylko przez jednego rodzica. Ten rodzic ma EBS i prawdopodobnie ma (lub miał) związane z nim objawy.
Nasilenie objawów EBS może być różne, od kruchości skóry i niewielkich pęcherzy na dłoniach i stopach, przez przypadki, w których pęcherze występują na całym ciele, po cięższe podtypy. Łagodne formy mogą ulec poprawie wraz z wiekiem, ale większość osób ma problemy z pęcherzami przez całe dzieciństwo i dorosłość.
Postać graniczna EB (EBJ)
Postać graniczna EB (JEB) stanowi około 10% wszystkich przypadków EB. W JEB oddzielenie naskórka i skóry właściwej następuje w błonie podstawnej. JEB jest dziedziczona po rodzicach jako cecha recesywna, co oznacza, że choroba rozwija się w wyniku odziedziczeniamutacji genu sprzężonego z EB od obojga rodziców. Rodzice prawdopodobnie nie mieli żadnych objawów EB.
Niektóre osoby dotknięte JEB będą miały „ciężką postać JEB” (wcześniej nazywany ciężkim uogólnionym JEB lub Herlitz JEB). U takich osób pęcherze występują na całym ciele, w tym na ustach, w nosie i gardle. Pęcherze te uniemożliwiają noworodkom prawidłowe odżywianie i oddychanie, powodując niedożywienie i niewydolność płuc. Niestety, ten ciężki podtyp JEB zagraża życiu i niewiele dzieci z tą postacią JEB żyje dłużej niż dwa lata.
Drugim głównym podtypem JEB jest „pośrednia postać JEB ” (wcześniej nazywany pośrednim uogólnionym JEB lub nie-Herlitz JEB). Objawy dla tej grupy są mniej poważne, ale nadal intensywne. Pęcherze pojawiają się na całym ciele, w tym w jamie ustnej, nosie i gardle. Niestety, śmiertelność jest ciągle dość wysoka w przypadku tej formy JEB. Osoby z pośrednią postacią JEB mogą dożyć dorosłości, ale często mają duże blizny, problemy z paznokciami i zębami, a także inne komplikacje ograniczające życie.
Dystroficzna postać EB
Dystroficzna postać EB (DEB) stanowi około 20% wszystkich przypadków EB. W DEB oddzielenie naskórka i skóry właściwej następuje pod błoną podstawną. DEB może być dziedziczona zarówno jako cecha dominująca, jak i recesywna, w zależności od mutacji genu sprzężonego z EB.
Nasilenie objawów jest bardzo różne w przypadku DEB. Wspólną cechą jest to, że pęcherze DEB prowadzą do powstawania blizn. W wielu przypadkach u osób z dominującą postacią DEB występują łagodne pęcherze i inne objawy, które w ograniczonym stopniu utrudniają życie tym osobom. Większość osób z recesywnym typem DEB musi stawiać czoła wielu innym ograniczeniom, ponieważ występujące u nich pęcherze i blizny są znacznie poważniejsze, a osoby te są narażone na zwiększone prawdopodobieństwo wystąpienia raka skóry. Dodatkowe objawy mogą obejmować oszpecenie w miarę gojenia się pęcherzy i ran; zrastanie się palców rąk i nóg, ograniczone ruchy stawów i zwężenie przełyku, co utrudnia połykanie.
Zespół Kindlera
Zespół Kindlera (KEB) to rzadki podtyp dziedzicznego EB, w którym pęcherze mogą występować na różnych warstwach skóry. KEB jest recesywną formą EB, co oznacza, że osoba dziedziczy mutację genu od obojga rodziców. Objawy mogą wahać się od łagodnych do ciężkich i obejmują kruchość skóry od urodzenia, wrażliwość na światło, przebarwienia i zgrubienia skóry. Choroba dziąseł, pęcherze w jamie ustnej i zapalenie jelit mogą wpływać na odżywianie, a osoby z KEB mają zwiększone ryzyko zachorowania na raka skóry.
Nasi partnerzy z Sieci Badawczej EB posiadają informacje na temat EB i jej klasyfikacji.
Chociaż pęcherze są objawem wszystkich rodzajów EB, nie są one spowodowane tym samym problemem. O tym gdzie, dlaczego i jak intensywnie pojawiają się pęcherze i odwarstwienie skóry decyduje to, w jakim genie związanym z EB występuje mutacja. Decyduje to również o tym, jaki typ EB ma dana osoba.
Obecnie istnieje 16 specyficznych genów w naszym DNA, które zostały powiązane z klasycznym EB. Mutacje w tych genach uniemożliwiają komórkom wytwarzanie funkcjonalnych białek, które utrzymują naskórek i skórę właściwą razem. Wiedza o tym, jaką rolę pełnią białka, które odpowiadają za utrzymanie skóry w całości, jest kluczowa dla zrozumienia, czym różnią się EBS, JEB i DEB. Pozwala również zrozumieć, dlaczego niektóre rodzaje EB są cięższe od innych i dlaczego leczenie jednego rodzaju EB może nie działać w przypadku wszystkich.
Poniżej znajdują się streszczenia, które opisują geny, kodowane przez nie białka i szczegóły dotyczące tego, gdzie i dlaczego dochodzi do odwarstwienia skóry w trzech głównych formach EB.
Postać zwykła EB
W EBS oddzielenie naskórka od skóry właściwej następuje w obrębie komórek podstawnych naskórka (Stratum basale), tuż nad błoną podstawną. Głównym problemem jest to, że białka łączące komórki podstawne z błoną podstawną nie są prawidłowo połączone z wewnętrznym strukturalnym elementem komórki (tzw. cytoszkieletem). Powoduje to rozerwanie i pęknięcie komórek podstawnych, i pozostawienie dolnej części tych komórek przytwierdzonych do błony podstawnej.
Najważniejsze białka i kodujące je geny podlegające mutacjom w EBS:
Keratyna typu 5 – [nazwa genu: KRT5]
Keratyna typu 14 – [nazwa genu: KRT14]
Plektyna – [nazwa genu: PLEC1]
Ilustracja 2: Diagram przedstawiający zdrową skórę i skórę ze zwykłą postacią EB.
Postać graniczna EB
W JEB oddzielenie naskórka od skóry właściwej następuje w błonie podstawnej, a konkretnie w podwarstwie nazywanej "blaszką jasną". Ta podwarstwa składa się z białek, które zakotwiczają komórki podstawne naskórka do skóry właściwej. Kiedy brakuje białek w tej warstwie lub nie są one funkcjonalne, ten punkt połączenia nie działa prawidłowo.
Podstawowe białka, których dotyczą mutacje genów w JEB to:
Kolagen typu XVII (znany również jako BPAG2) - [Nazwa genu: COL17A1]
Laminina-332 (znana również jako Laminina-5) - [Nazwy genów: LAMA3, LAMB3 i LAMC2]
Ilustracja 3: Diagram przedstawiający zdrową skórę i skórę z graniczną postacią EB.
Dystroficzna postać EB
W DEB oddzielenie naskórka od skóry właściwej następuje pod błoną podstawną. Najważniejszym białkiem wiążącym błonę podstawną ze skórą właściwą jest kolagen VII. Białko to tworzy fibryle kotwiczące, które wplatają się w białka strukturalne skóry właściwej. Bez kolagenu VII błona podstawna łatwo odrywa się od skóry właściwej.
Podstawowym białkiem zmienionym na skutek mutacji genu w DEB jest:
Kolagen typu VII - [nazwa genu: COL7A1].
Ilustracja 4: Diagram przedstawiający zdrową skórę i skórę z dystroficzną postacią EB.
Zespół Kindlera
KEB może dotyczyć różnych i wielu warstw skóry. Kiedy brakuje określonego białka lub nie jest ono funkcjonalne, zaburza to zdolność keratynocytów (komórek skóry) do normalnego wzrostu i podziału oraz do łączenia naskórka ze skórą właściwą.
Podstawowym białkiem zmienionym na skutek mutacji genu w KEB jest:
Kindlina-1 - [nazwa genu: FERMT1].
Leczenie EB
Obecnie nie ma zatwierdzonych żadnych metod leczenia EB, które korygowałyby leżące u podstaw EB mutacje genetyczne. Leczenie koncentruje się przede wszystkim na zapobieganiu wystąpienia lub łagodzeniu objawów EB. Środki zapobiegawcze mogą obejmować stosowanie okładów i podkładów w celu zminimalizowania tarcia, uderzeń i drobnych urazów skóry. Celem leczenia może być również zapobieganie infekcjom oraz pomoc w gojeniu się pęcherzy i owrzodzeń.
W cięższych przypadkach EB leki mogą być stosowane w celu zapobiegania zakażeniom otwartych ran i owrzodzeń. Można również przeprowadzić operację, aby zapobiec zrastaniu się palców u rąk i nóg lub nadmiernemu zwężeniu gardła i przełyku (przewodu pokarmowego).
W skrajnych przypadkach EB, lekarze mogą pokryć duże otwarte rany poprzez przeprowadzenie przeszczepów skóry. Przeszczepy skóry od dawców są jednak bardzo rzadkie, ponieważ wiadomo, że układ odpornościowy pacjenta prawie zawsze je odrzuci, nawet jeśli pacjent i dawca są ściśle dopasowani. W związku z tym lekarze często stosują tymczasowe substytuty skóry. Coraz powszechniejsze stają się również nowe "biologiczne substytuty skóry".
Obecne badania
Poprawa jakości życia
Obecnie nie istnieją klinicznie zatwierdzone terapie „naprawiające” przyczyny EB, czyli mutacje genetyczne. Wielu badaczy, firmy farmaceutyczne i zajmujące się zaopatrzeniem medycznym, pracują nad opracowaniem lepszych produktów w celu łagodzenia objawów EB.
Wśród testowanych produktów są bandaże i okłady, jak również maści i balsamy wspomagające gojenie się skóry.
Obecnie prowadzonych jest kilka prób klinicznych z wykorzystaniem różnych rodzajów opatrunków na rany i leków w kremach do stosowania miejscowego, które potencjalnie mogłyby przyspieszyć proces gojenia się skóry u osób z EB.
W ciężkich przypadkach EB konieczne może okazać się zasklepienie obszernych, otwartych ran przez lekarza. Często obejmuje to pokrywanie ran syntetycznymi substytutami skóry. Ostatnio firmy medyczne oferują jednak bardziej zaawansowane „biologiczne substytuty skóry”, takie jak Biobrane®, Integra®, Orcel®, Apligraft® i inne. Celem tych zaawansowanych substytutów skóry jest poprawa gojenia ran przez wykorzystanie syntetycznych siatek, na których komórki efektywnie rosną, białek obecnych w skórze, a czasami nawet żywych keratynocytów (komórek skóry).
Wymienione metody stanowią jedynie nieliczne przykłady nowych technologii i produktów, które są opracowywane, aby znacznie poprawić jakość życia osób dotkniętych EB.
Tworzenie „nowej” skóry
Idealną metodą leczenia zaburzeń genetycznych jest naprawa i zastąpienie zmutowanego genu w DNA chorego. Badacze tworzą „terapie genowe”, które działają właśnie w ten sposób. W ciągu ostatnich lat pojawiło się kilka skutecznych badań klinicznych i terapii. Te obiecujące wyniki wskazują, że skuteczne leczenie może nadejść. Niedługo, po raz pierwszy mogą być dostępne terapie leczące przyczyny EB.
Terapie genowe są obecnie na wczesnym etapie rozwoju. Nowe odkrycia i technologie, takie jak edycja genów metodą CRISPR-Cas9, niezwykle przyspieszają edytowanie DNA i genów w komórkach. Zmiana DNA w komórkach danej osoby nie jest jednak łatwym zadaniem. Proces ten może wiązać się z poważnym ryzykiem. Jeśli edycja bądź wprowadzanie nowych genów nie przebiegnie prawidłowo, może dojść do rozwoju nowotworu, powstania komplikacji i pogorszenia kondycji pacjenta. Badania kliniczne są kluczowe dla identyfikacji ryzyka związanego z nową terapią i weryfikacji jej skuteczności.
W wielu przypadkach w terapiach genowych rozważa się wykorzystanie komórek macierzystych . W przypadku terapii EB, badacze chcą edytować DNA komórek macierzystych skóry, zamiast modyfikować DNA innych typów komórek skóry. Jest to spowodowane faktem, że dodanie (lub edycja) genu w komórce macierzystej skóry spowoduje jego przeniesienie do wszystkich komórek naskórka, które powstaną z komórki macierzystej. To bardzo ważne ze względu na fakt, że niemal wszystkie keratynocyty w naszej skórze są wymieniane co 4-5 tygodni. Dodatkowo, komórki macierzyste skóry stale odtwarzają swoją populację, zatem każda zmiana DNA wprowadzona do komórki macierzystej może potencjalnie być „trwała” (lub przynajmniej będzie utrzymywać się tak długo, jak komórka macierzysta przeżyje).
Kilka terapii EB, które aktualnie są na etapie badań klinicznych, sprawdzają metody edycji DNA komórek macierzystych skóry pacjentów, w celu wyhodowania „nowej” zdrowej skóry do przeszczepu. Pomimo różnic w metodach i szczegółach między poszczególnymi próbami klinicznymi, zamysł ogólny pozostaje taki sam. Obejmuje on pobranie próbki skóry od osoby cierpiącej na EB i hodowanie wyizolowanych z niej komórek w laboratorium, z nadzieją, że w tych próbkach znajdują się komórki macierzyste skóry. Następnie stosuje się techniki edytowania DNA obejmujące korektę genów lub wprowadzenie nowych genów do komórek i wznowienie produkcji prawidłowych białek. Każda komórka z nowym zmodyfikowanym DNA jest następnie hodowana w laboratorium z wykorzystaniem zaawansowanych metod, w celu uzyskania warstw skóry. W kolejnych etapach wyhodowane laboratoryjnie warstwy skóry mogą być przeszczepione pacjentowi. Niektóre z tych podejść obejmują wykorzystanie indukowanych pluripotencjalnych komórek macierzystych (iPSC), rodzaju komórek macierzystych, które mogą być otrzymane z komórek naszego ciała. Te komórki mogą dzielić się w nieskończoność i różnicować w każdy rodzaj komórek występujących w skórze. Mogą być zatem znakomitym źródłem komórek do zastąpienia uszkodzonej tkanki. Pomimo dopuszczenia możliwości wykorzystania indukowanych pluripotencjalnych komórek macierzystych (iPSC) w badaniach klinicznych dotyczących różnych chorób, pacjenci, którzy przyjmują przeszczep z komórkami wywodzącymi się od iPSC, mogą być narażeni na większe ryzyko rozwoju nowotworu.
Opisywany rodzaj leczenia wymaga czasu i pracy, aby zidentyfikować genetyczną przyczynę EB u każdej osoby dotkniętej tą chorobą, opracować specyficzne podejście do edycji DNA, przeprowadzić wielotygodniową hodowlę komórek, a następnie przeprowadzić przeszczep skóry. Wymaga on również specjalistycznych obiektów badawczych i narzędzi do hodowli komórek. Pomimo tych przeszkód, odnotowano znaczne postępy pokazujące że takie terapie mogą działać.
W następnej sekcji omówiono jedną z takich pomyślnych historii.
Linki do stron internetowych użytych w tekscie (dostępne w języku angielskim):
Badania europejskie w centrum uwagi: historia sukcesu
W 2017 roku grupa badaczy i lekarzy kierowana przez profesora Michele De Luca doniosła o skutecznym wykorzystaniu terapii genowej i wyhodowanych laboratoryjnie przeszczepów skóry, w celu uratowania chłopca, któremu brakowało ponad 80% skóry. Chłopiec chorował na JEB spowodowane mutacją w genie LAMB3, którą odziedziczył od obojga rodziców. Wszystkie zastosowane metody leczeniazawiodły i lekarze obawiali się, że chłopiec nie przeżyje. Władze rządowe udzieliły pozwolenia na „humanitarne zastosowanie” terapii genowej, która do tamtej pory była stosowana tylko dwa razy u jednego pacjenta. Rodzice chłopca również wyrazili zgodę na terapię, mimo informacji, że może być ona śmiertelna.
Badacze pobrali próbki skóry chłopca i hodowali jego komórki w laboratorium. Mieli nadzieję na uzyskanie skupiska komórek zawierającego komórki macierzyste skóry. Następnie, zastosowali wektor wirusowy w celu wprowadzenia funkcjonalnej kopii genu LAMB3 do komórek macierzystych skóry. Taka procedura nie naprawiała mutacji, jaką chłopiec miał w swoich genach lecz wprowadzała nową kopię genu LAMB3 do komórek macierzystych skóry chłopca. Nowy, prawidłowy gen pozwolił komórkom na wytwarzanie funkcjonalnego białka - lamininy. Komórki macierzyste skóry z dodanym, funkcjonalnym genem LAMB3 były następnie użyte do wyhodowania płatów skóry, z wykorzystaniem zaawansowanych technik hodowli. Ten proces trwał kilka tygodni. Chłopiec był poddany dwóm przeszczepom fragmentów wyhodowanej w laboratorium skóry, w odstępie miesiąca. Ostatecznie chłopiec przeżył a 80% powierzchni jego ciała pokrywa skóra zbudowana z komórek ze zmodyfikowanym DNA. W ocenie badaczy skóra ta pod wieloma względami przypomina normalną skórę.
Efekt tego zabiegu przekroczył oczekiwania. Niestety podobne terapie genowe nie są jeszcze zatwierdzone klinicznie ani powszechnie dostępne. Należy również pamiętać, że docelowo terapie genowe muszą „korygować” mutacje w genach. Taka spersonalizowana medycyna jest droga, wymaga specjalnych obiektów i współpracy wielu ludzi. Miejmy nadzieję, że takie terapie będą kliniczne zatwierdzone, przystępne i powszechne w leczeniu wielu form EB.
Linki do stron internetowych użytych w tej sekcji (dostępne w języku angielskim):
Przyszłość wygląda obiecująco. Sukces wyżej opisanego zabiegu, jak również pozytywne wyniki innych badań doprowadziły do rozpoczęcia kilku nowych badań klinicznych. M. De Luca z Centrum Medycyny Regeneracyjnej „Stefano Ferrari” w Modenie we Włoszech, razem z JW. Bauer’em z Uniwersyteckiego Szpitala Dermatologicznego w Salzburgu w Austrii kierują kilkoma badaniami klinicznymi wykorzystującymi podobne, jednak bardziej zaawansowane metody, w celu zastąpienia mutacji w genach COL7A1 i COL17A1 (Badanie 1, Badanie 2). Badacze z innych ośrodków naukowych również opracowują własne terapie genowe, takie jak w badaniu klinicznym kierowanym przez Jean Yuh Tang z Uniwersytetu Medycznego Stanford, które jest również ukierunkowane na badanie mutacji w genie COL7A1, leżącej u podstaw DEB.
Miejmy nadzieję, że badacze opracują bezpieczne, skuteczne i przystępne terapie leczenia EB.
Inne podejścia terapeutyczne
Jak wspomniano w poprzedniej sekcji, przygotowanie skóry ze zmienionym DNA wiąże się z poważnymi wyzwaniami; procedura jest złożona, droga i ryzykowna. Mogą upłynąć lata zanim takie podejście stanie się osiągalną metodą leczenia.
Te wyzwania skłoniły naukowców do myślenia o innych możliwościach leczenia EB, które byłyby szybsze, zatwierdzone klinicznie i dostępne finansowo dla chorych dotkniętych EB. Obejmują one komórki macierzyste, leki, ekstrakty roślinne i wiele innych terapii. Poniżej przedstawiono przykłady dwóch z nich.
Terapeutyczne kremy i balsamy do stosowania miejscowego są opracowywane w celu przeciwdziałania problemom spowodowanym przez specyficzne mutacje EB. Dwa szczególnie interesujące badania kliniczne obejmują wykorzystanie kremów, których zadaniem jest naprawa, bądź zastąpienie genu COL7A1, co może pomóc pacjentom z DEB. Jeden z kremów zawiera składniki stymulujące komórki skóry do pominięcia mutacji w genie COL7A1 podczas wytwarzania kolagenu VII. Drugi krem zawiera natomiast cząsteczki wirusowe wprowadzające prawidłową kopię genu COL7A1 do komórek. (Te cząsteczki wirusowe nie mogą się namnażać, dzięki czemu są bezpieczne do stosowania.)
Przeszczepy szpiku kostnego są zazwyczaj kojarzone z białaczką i innymi chorobami krwi i układu odpornościowego. Niektóre grupy badawcze testują jednak terapię EB obejmującą allogeniczne przeszczepy szpiku kostnego, w których komórki macierzyste pochodzą od zdrowych dawców. Przewagą takiego podejścia jest to, że przeszczepy szpiku kostnego są skutecznie wykonywane od wielu lat, a technika ich wykonywania została przez ten czas udoskonalona. Wstępne wyniki sugerują, że zdrowe komórki macierzyste szpiku kostnego (lub ich komórki potomne) produkują białka tkanki łącznej, takie jak kolagen VII, które nie występują u jednostek z DEB. Metoda ta wiąże się z ograniczeniami, takimi jak konieczność znalezienia dawcy dopasowanego immunologicznie, a także z wysokim ryzykiem powikłań.
Bardziej obszerna lista badań klinicznych w terapii EB jest dostępna na stronie clinicaltrials.gov. (Proszę pamiętać, że na tej stronie internetowej są jedynie wymienione badania kliniczne. Nie jest potwierdzone czy są one bezpieczne, naukowo uzasadnione lub prowadzone przez renomowane instytucje. Prosimy o przeczytanie wszelkich zastrzeżeń, rozmowę z zaufanymi dostawcami usług medycznych i zapoznanie się z ryzykiem i potencjalnymi korzyściami.)
Linki do stron internetowych użytych w tej sekcji (dostępne w języku angielskim):
Stanford University EB Clinic – informacje izasobyo EB, takie jak podstawowe wskazówki dotyczące opieki nad osobami z EB oraz linki do filmów na temat pielęgnacji ran